Issuing authorization for a clinical trial of a medicinal product – practical tips under EU and Polish law
Publication date: February 26, 2024
Given the current medical standards, the birth of a new medicinal product (drug) can only become a fact as part of a long-term and expensive research and development process. Such a process carries a high risk of failure. Typically, the research and development process takes many years and its effectiveness is low. However, such research is undoubtedly needed.
How to obtain authorization for a clinical trial of a medicinal product?
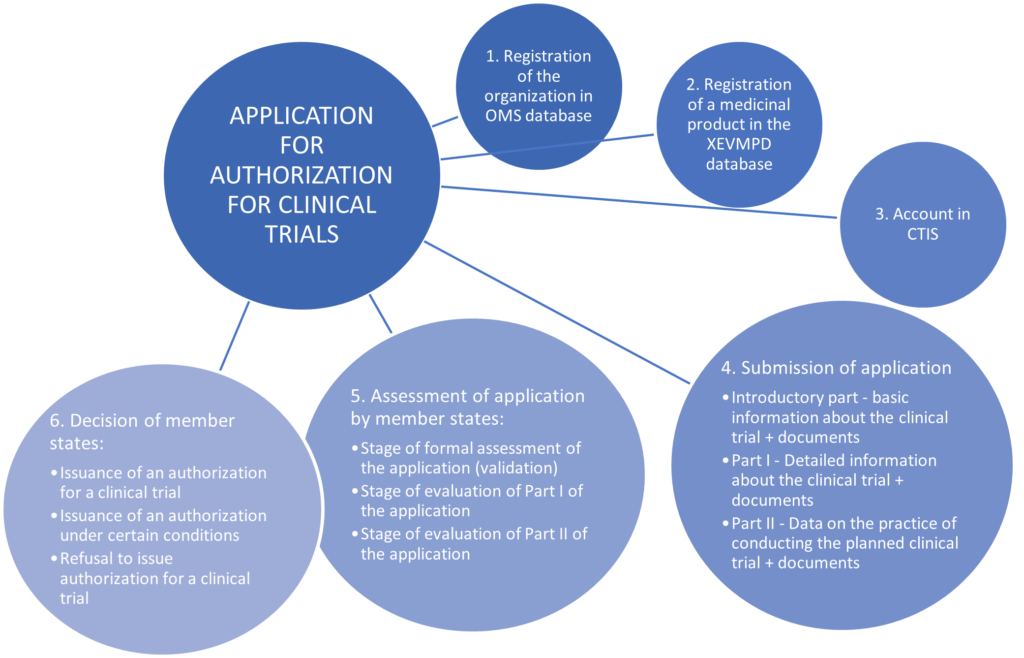
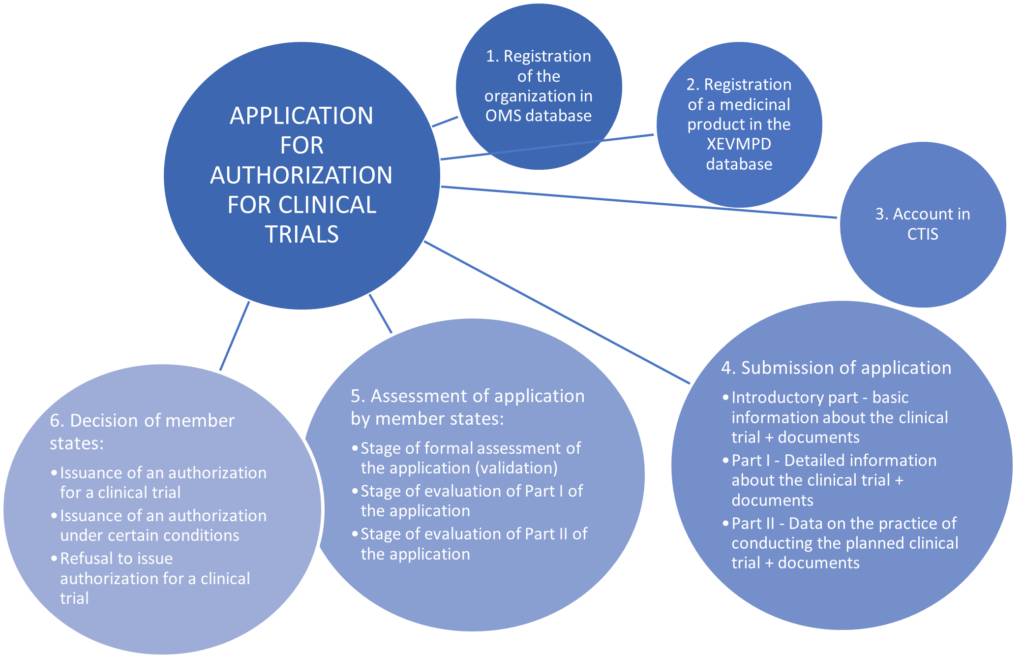
To obtain permission to conduct such a study in the territory of the Republic of Poland, there must be first submitted an application for authorization. Such an application may only be submitted by the sponsor – which is understood as “an individual, company, institution or organisation which takes responsibility for the initiation, for the management and for setting up the financing of the clinical trial” (Article 2(2)(14) of Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC). The sponsor is obliged to submit such an application via CTIS (i.e. the new European system, Clinical Trials Information System).
From January 31, 2022, the Clinical Trials Regulations (CTR) applies, harmonizing the processes for submitting applications, assessing and supervising clinical trials in the European Union. The core of the changes introduced by CTR is the new Clinical Trials Information System (CTIS). CTIS is a single point of access for clinical trial sponsors and regulators to submit and evaluate clinical trial data, which includes a publicly accessible, searchable database for healthcare professionals, patients and the general public.
In the past, sponsors had to submit clinical trial applications separately to the national competent authorities and ethics committees in each country to obtain regulatory approval to conduct a clinical trial, and registration and publication of results were also separate processes. With CTIS, sponsors can now apply for authorizations in up to 30 European Union and European Economic Area (EEA) countries at the same time and with the same documentation. The publication of information about the study is built into the system.
The use of CTR and the launch of CTIS – in EU and EEA countries (Iceland, Liechtenstein and Norway) – strengthens Europe’s position as an attractive location for clinical trials. The new regulation improves the application, supervision and public registration of clinical trials: all clinical trial sponsors will use the same system (CTIS) and follow the same process for applying for clinical trial authorization, regardless of where they are based and which national authority or bioethics commission they are dealing with. The new system has a dedicated secure workspace for research sponsors where they can submit and manage their clinical trial requests. There is a similar secure workspace for authorizing authorities who can easily interact with the sponsor and quickly collaborate and exchange information with other authorities.
Due to the fact that transparency is a core feature of CTR, CTIS also includes a searchable public website that will prospectively include detailed information and results of all clinical trials authorized through the system.
The CTR provides for a three-year transition period. Member States will work in CTIS as soon as the system is launched. For one year, until January 31, 2023, clinical trial sponsors could still decide whether to submit an initial clinical trial application under the previous system (Clinical Trials Directive) or via CTIS. From 31 January 2023, the submission of initial applications for clinical trials via CTIS became mandatory, and by 31 January 2025, all ongoing trials approved under the Clinical Trials Directive will be subject to the new regulation and will need to be transferred to CTIS.
EU and EEA Member States are responsible for the authorization and supervision of clinical trials, while the European Medicines Agency (EMA) is responsible for maintaining CTIS. The European Commission (EC) supervises the implementation of the Clinical Trials Regulation.
CTIS account
In accordance with the CTR (Regulation (EU) No. 536/2014 of the European Parliament and of the Council of 16 April 2014 repealing Directive 2001/20/EC, applications for authorization to conduct clinical trials can only be submitted through the new European CTIS system, so it is advisable to create an account in this system. The user of this system may be the sponsor, an authorized employee or associate of the sponsor. In order to gain access to sponsor panel in CTIS, the user must have a European Medicines Agency account). If they have previously created an account in one of the other EMA applications (e.g. Eudralink, SPOR, IRIS, EudraVigilance, OMS), they can use the same EMA account to also have access to the sponsor panel in CTIS. However, if the user does not have any EMA account, it is necessary to create an account by registering on the EMA account management website. When registering in CTIS, there are several options and depending on which ones the sponsor chooses, it may also turn out that it will be necessary to establish and assign appropriate access roles to users allowing them to submit an application for permission to conduct the study on behalf of the sponsor (authorized employees and associates of the sponsor).
The European Medicines Agency recommends that sponsors carefully read the CTIS training modules and the CTIS Sponsor Handbook, which contains detailed information on how to use the European Clinical Trials Program, before creating an account and submitting an application.
Organization registration
Before a sponsor submits an application, they must be sure that their organization is registered with the EU’s Organization Management Service (OMS). This is a necessary condition for submitting an application for authorization to start clinical trials.
Registration of a medicinal product
Before submitting an application for authorization to conduct clinical trials, the sponsor must not forget to register (or ensure that they are registered) details of all medicinal products that will be used in the clinical trial in eXtended EudraVigilance Medicinal Product Dictionary (XEVMPD). The XEVMPD database should contain data on all medicinal products authorized for marketing in the EEA or under development (including at the clinical trial stage). However, there is no need to register the so-called placebo in the database XEVMP.
Documentation and data needed to submit an application
Submitting an application in the CTIS system is divided into three parts: preliminary part, part I and part II.
Introductory part
At this stage of submitting an application in the system, there must be provided the simplest, basic information regarding the planned clinical trial. First of all, it concerns:
- name of the study and study sponsor (selected from the OMS list).
There is also needed the submission of documents such as:
- cover letter with letter B of Annex I to the CTR;
- confirmation of payment of the fee (Details regarding the liability to the fee and its amount in the Republic of Poland are specified in Article 58 of the Clinical Trials Act);
- declaration of compliance with GDPR requirements.
This section should also include any interested Member States and propose which of them will be the reporting Member State (The sponsor shall propose one of the Member States concerned as reporting Member State). When selecting Member States, one should also specify how many expected participants from a given country will take part in the study.
Part I
At this stage there needs to be provided more detailed information, such as:
- Phase of the planned clinical trial;
- The type of disease to be examined;
- Therapeutic area;
- Scope and purpose of the clinical trial;
- Criteria for inclusion and exclusion from the clinical trial;
- Study endpoints;
- Duration of a clinical trial from the beginning of recruitment to its end;
- Data regarding the recruited participants of the clinical trial (age range, gender, whether they are healthy people or whether they have any diseases).
One must also not forget about the documents at this stage:
- Test protocol in accordance with point D of Annex I to the CTR;
- Investigator’s brochure in accordance with point E of Annex I to the CTR;
- Documentation regarding good manufacturing practicein relation to the medicinal product under trial in accordance with point F of Annex I to the CTR;
- Auxiliary medicinal product dossier in accordance with point G of Annex I to the CTR;
- Scientific indications and clinical investigation plan in the pediatric population in accordance with point I of Annex I to the CTR (if applicable);
- Content of the labeling of the investigational medicinal product in accordance with Annex VI to the CTR.
Part II
As part of this stage, data regarding the practice of conducting the planned clinical trial should be provided. Mainly such as:
- Identification of clinical trial sites and investigators for each Member State concerned.
Of course, as in the previous stages, we cannot forget about the documents:
- Rules for recruitment to a clinical trial in accordance with point K of Annex I to the CTR;
- Information for participants of the clinical trial;
- Informed consent form and informed consent procedure in accordance with point L of Annex I to the CTR;
- Documents confirming the appropriate qualifications of the researcher in accordance with point M of Annex I to the CTR;
- Documents confirming the appropriate quality of selected clinical trial centers in accordance with point N of Annex I to the CTR;
- Proof of insurance or membership in a national compensation mechanism in accordance with the national law of the Member State concerned in accordance with point O of Annex I to the CTR;
- Documents regarding the financing of the clinical trial in accordance with point P of Annex I to the CTR;
- Documents regarding compliance with the personal data protection rules in the Member State in accordance with point R of Annex I to the CTR;
- Documents confirming compliance with the rules regarding the collection of biological samples from participants and the storage and future use of these samples.
Language of the documentation submitted
Article 10 of the Polish Clinical Trials Act contains detailed requirements regarding the language in which the documentation described above should be submitted. Pursuant to this article, most documents attached under stage I may be prepared in English or Polish, while the vast majority of documents submitted under part II must be prepared in Polish.
The sponsor’s choice as to how to submit the application
The sponsor may submit the full application, which will consist of both Part I and Part II, simultaneously to all interested Member States. He may also submit a partial application: submit Part I to all interested Member States, and Part II not to any Member State or only to some Member States)
The role of the reporting Member State and the assessment of the proposal by Member States
In the initial part of submitting the application, the sponsor is obliged to propose which of the selected interested Member States should act as the reporting Member State. This proposal is not binding – this means that the Member States concerned may, by agreement, choose another Member State as a reporting Member State. In the event that countries cannot agree on the choice of reporting Member State, the country proposed by the sponsor shall become the reporting Member State. However, if a clinical trial concerns only one Member State, that Member State becomes the reporting Member State. After determining the Member State acting as the reporting Member State, countries proceed to the evaluation of the application, which takes place in three main stages: the stage of formal evaluation of the application, the stage of evaluation of Part I of the application, the stage of evaluation of Part II of the application.
Validation, i.e. the stage of formal assessment of the application
Within 10 days of submission of the application, the reporting Member State shall validate the application:
- It takes into account the comments submitted by the other Member States concerned;
- Provides the sponsor with information via CTIS whether the clinical trial to which the application relates falls within the scope of the CTR;
- Checks whether the application documentation is complete in accordance with Annex I to the CTR.
Where the reporting Member State has not provided the sponsor with information in the above-mentioned deadline, the clinical trial to which the application relates is considered to be within the scope of the CTR and the application documentation is considered complete.
Where the reporting Member State finds that the application dossier is incomplete or that the clinical trial covered by the application is not covered by the CTR, it shall inform the sponsor thereof via CTIS and set a deadline of no more than 10 days for the sponsor to submit comments on the application or supplementing the application documentation in CTIS.
Within 5 days of receiving comments on the application or completed application documentation, the reporting Member State notifies the sponsor whether the application complies with the CTR requirements. Where the sponsor does not complete the application within the deadline, the application will be deemed to have expired in all Member States concerned.
Stage of evaluation of Part I of the application
At this stage, the reporting Member State assesses the application in terms of the following aspects:
- Whether the clinical trial is low-intervention, where the sponsor has identified it as such;
- Compliance with Chapter V of the CTR in relation to:
– anticipated therapeutic and public health benefits;
– risks and inconvenience for the participant;
- Compliance with the requirements set out in Chapter IX of the CTR for the manufacture and import of investigational medicinal products and auxiliary medicinal products;
- Compliance with the labeling requirements set out in Chapter X of the CTR;
- Completeness and adequacy of the investigator’s brochure.
When the above-mentioned aspects pass the verification, the reporting Member State shall draw up an evaluation report which shall contain one of the following conclusions:
- Conducting the clinical trial is permissible in accordance with CTR;
- Conducting the clinical trial is permissible in accordance with the CTR, after meeting the additional requirements listed in the conclusion;
- Conducting the clinical trial is unacceptable according to the CTR.
Within 45 days from the date of completion of the formal assessment stage of the application (validation date), the reporting Member State submits the final version of Part I of the assessment report to the sponsor via CTIS. If it wishes to consult experts, the reporting Member State may extend the deadline by a further 50 days in the case of clinical trials concerning advanced therapy investigational medicinal products or medicinal products as defined in point 1 of the Annex to Regulation (EC) No 726/2004 of the Parliament European Union and of the Council of 31/03/2004 establishing Community procedures for the authorization and supervision of medicinal products for human and veterinary use and establishing the European Medicines Agency.
In the period from the validation date to the report submission date, the reporting Member State may request additional information from the sponsor (CTIS). The sponsor must provide the requested information within the deadline specified by the reporting Member State. This deadline may not exceed 12 days from receipt of the application. In order to obtain and assess additional information from the sponsor, the Member State may extend the deadline by a maximum of a further 31 days.
Stage of evaluation of Part II of the application
The assessment of Part II of the application differs in that it is not carried out by the reporting Member State, but by each interested Member State separately. Member States shall assess the application on the aspects set out in Article 7 section 1 of CTR, in particular:
- compliance of the principles of remuneration or compensation for research participants with the requirements set out in Chapter V of the CTR;
- whether researchers are appropriately qualified under national law;
- whether the centers where the research is to be carried out are suitable for carrying out the research in accordance with the CTR requirements.
Each interested Member State shall assess Part II of the application within 45 days of the validation date (date of completion of the formal assessment stage) and submit Part II assessment reports to the sponsor via CTIS. The evaluating Member States may also make requests for additional information (CTIS) from the sponsor. The sponsor should provide additional information via the CTIS system within the deadline specified by the Member State, which may not exceed 12 days. After receiving the additional information, the Member State concerned shall evaluate it within a period not exceeding 19 days.
Decision of the Member States on the clinical trial
Each Member State concerned shall notify the sponsor via CTIS whether:
- it issues authorization for a clinical trial;
- it grants authorization under certain conditions;
- refuses to issue the authorization.
Notification is made in a single decision within 5 days from the date of submission of Part I of the assessment report or from the last day of assessment of Part II of the application, whichever is later. In principle, Part I of the assessment report, prepared by the reporting Member State, is binding on the other Member States. A non-reporting Member State may challenge the positive conclusion of a Reporting Member State in respect of Part I of the report only on the following grounds:
- if that State considers that participation in a clinical trial would result in the subject receiving treatment that is inferior to that which constitutes standard clinical practice in the Member State concerned;
- medicinal products whose use is prohibited or restricted in accordance with national law are to be used in a clinical trial (Article 90 of the CTR);
- under additional considerations regarding participant safety and the robustness and reliability of the data raised in the Part I evaluation.
Refusal to issue the authorization
Where a Member State does not agree with the conclusion regarding Part I, it shall inform the Commission, all Member States and the sponsor of its disagreement via CTIS, together with detailed reasons.
The Member State concerned may refuse to authorize clinical trials if it disagrees with the conclusion of the reporting Member State in relation to Part I of the assessment report or if, on a duly substantiated basis, it finds a lack of compliance with the aspects covered in Part II of the assessment report or if the ethics committee issued a negative opinion. Each Member State is obliged to put in place an appeals procedure in the event of such a refusal.
If the Member State concerned has not notified the sponsor of its decision within the deadline referred to above (5 days from the date of submission of Part I of the assessment report or from the last day of assessment of Part II of the application, whichever is later), the conclusion on Part I of the assessment report is deemed to be the decision of the Member State concerned on the application for authorization of the clinical trial.
Withdrawal of the application by the sponsor
The sponsor has the right to withdraw its application at any time up to the date of submission of the report. Where an application is withdrawn before the sponsor has been informed of the conclusion on Part I of the assessment report, the withdrawal shall apply to the entire application in all Member States concerned. After withdrawing the application, one can resubmit it. Such a resubmitted application is considered a new application for authorization of another clinical trial.
Sponsors’ caution
Sponsors must also pay attention to the special regulation contained in Art. 8 section 9 of CTR: if no subject has been enrolled in a clinical trial in the Member State concerned within 2 years from the date of notification of the authorization, the authorization shall expire in that Member State concerned, unless, at the request of the sponsor, this period has been extended in accordance with the procedure set out in Chapter III of CTR.
A chart showing the clinical trial authorization application process