New transparency requirement for public financial support received for drug research and development
Publication date: March 11, 2026
Medicines resulting from research and development (R&D) activities include medicinal products developed through a multi-stage process of preclinical and clinical trials conducted to demonstrate their quality, safety, and efficacy. This process is lengthy, expensive, and fraught with a high risk of failure. It is financed by both private and public funds (according to research by Claudie Wild, Ozren Sehic, Louise Schmidt, and Daniel Fabian, “Public contributions to R&D of medical innovations: A framework for analysis,” 26 publications were identified, which found that half of all approved medicines and >90% of target medicines are linked to public sector institutions or their funding), including EU funds and national innovation support programs. From a pharmaceutical law perspective, R&D medicines are subject to specific regulatory requirements at all stages of the product lifecycle, from clinical trials to marketing authorization and pharmacovigilance. In this context, the proposed changes to EU pharmaceutical regulations, concerning data standardization and information transparency, constitute part of a broader regulatory framework for the functioning of the innovative medicines market. The research and development process is as follows:
The scope of the European Medicines Agency (EMA) is crucial to the transparency of the EU’s regulatory system for medicinal products. It is the central entity responsible for, among other things, the evaluation, monitoring, and safety supervision of medicines. The EMA collects extensive scientific, clinical, and pharmacovigilance data from across the European Union and then makes it available in a structured and comparable format. Publishing assessment reports, summaries of regulatory decisions, and information on adverse reactions increases the transparency of the decision-making process and allows for verification of its merits by the scientific community and the public. Such transparency fosters public trust in regulatory institutions, reduces the risk of arbitrary decisions, and strengthens the legitimacy of the healthcare system by demonstrating that decisions regarding the approval and supervision of medicines are based on specific data and established procedures.
Under current law, Article 57 of Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency is of particular importance. Article 57, which sets out the tasks and competences of the European Medicines Agency (EMA), is responsible for the scientific assessment of the quality, safety, and efficacy of medicines for humans and animals, coordinates the procedures for authorizing medicines to be placed on the market in the EU, conducts pharmacovigilance, collects and makes available information on medicines (including public databases), supports Member States, EU institutions, and marketing authorisation holders, and ensures cooperation and exchange of information at the EU and international levels.
The new transparency requirement regarding public financial support for research and development activities in the field of medicinal products aims to increase transparency in the drug development and market launch process and to enable the assessment of the impact of public funds on their development and commercialization. The essence of this obligation is to disclose information about direct and – in certain cases – indirect financial support from public or quasi-public funds, in particular EU funds, national research programs, grants, subsidies, reliefs, or other financing mechanisms that may have had a significant impact on the research, development, manufacturing, or marketing authorization of a medicinal product.
Currently, the EMA provides a wide range of information on medicinal products through public databases such as European Public Assessment Reports (EPARs) and clinical trial registries, which ensure transparency in the evaluation process, safety, and efficacy of medicines. However, the scope of data disclosed focuses primarily on scientific and regulatory aspects—including clinical trial results, therapeutic indications, approval decisions, and risk information—without systematically including financial data related to research and development (R&D). In this context, the EMA could serve as a potential information provider, integrating existing regulatory resources with data on the funding sources for pharmaceutical innovation. This would foster greater transparency, a better understanding of the contribution of public and private funds to drug development, and strengthen stakeholder confidence in the European regulatory system.
In response to the current state of the pharmaceutical market, on 23 July 2025, an implementing regulation was published in the Official Journal of the EU, amending Implementing Regulation (EU) No 520/2012 on the performance of pharmacovigilance activities provided for in Regulation (EC) No 726/2004 of the European Parliament and of the Council and Directive 2001/83/EC of the European Parliament and of the Council. Article 57 of the proposed directive aims to ensure transparency regarding public and quasi-public financial support for activities aimed at researching and developing medicines. This is to be achieved by disclosing direct financial support that may have influenced the development and commercialization of these products.
The disclosure obligation is addressed in particular to marketing authorization holders and other entities responsible for conducting research and product development, including clinical trial sponsors, entities with capital or organizational ties, and – to the extent they participate in the R&D process – research and academic institutions working in collaboration with the pharmaceutical industry. This obligation covers both support received directly by the entity and funds transferred through consortia, public-private partnerships, or other collaborative structures.
Disclosure of information should occur at a specific stage in the development cycle of a medicinal product, in particular in connection with the marketing authorization procedure, its amendments, or updates to the regulatory documentation, and, where appropriate, during post-market surveillance. The form of data disclosure should ensure its accessibility, comparability, and comprehensibility for regulatory authorities and the public, while respecting commercial confidentiality and the protection of sensitive data. As a general rule, this information should be provided through central EU systems and registries, allowing for public consultation or, in the case of restricted data, access by the relevant supervisory authorities.
Transparency in drug R&D funding is not indifferent to the pharmaceutical market; it can be crucial in the context of pricing and reimbursement negotiations. Disclosure of information on public financial support received for drug development, as provided for in the proposed EU legislation, can help assess the true costs of innovation and the contribution of public funds to its development. Increased transparency can foster a more balanced relationship between public funding and the commercialization of innovations, and can facilitate social access to the results of publicly funded research.
Amendments to Article 26 of the Implementing Regulation clarify the electronic formats and standards used to submit information on medicinal products authorized for marketing in the Union, in particular by referencing the XEVPRM Communication and IDMP standards, in line with data published by the European Medicines Agency pursuant to Article 57(2) of Regulation (EC) No 726/2004. This regulation strengthens the EMA’s role as the entity responsible for maintaining reference datasets on medicinal products, while the scope of information submitted remains formally linked to regulatory, procedural, and pharmacovigilance data. Article 57 of Regulation 726/2004 specifies the Agency’s tasks primarily in the area of assessing the quality, safety, and efficacy of medicinal products and collecting and publicly making information in this regard available, including by maintaining an EU-wide database of medicinal products accessible to the public.
The new wording of Article 26 does not introduce separate substantive competences for the EMA, but rather refers to the Agency’s existing obligations regarding the publication and standardization of data on medicinal products. In this sense, it is possible to use existing IT systems and databases referred to in Article 57 as a reference point for other information obligations under EU law, as long as they remain based on the same product identifiers and data structures. However, this regulation does not prejudge the scope or nature of information beyond strictly regulatory data, including information on research and development funding, which may be subject to separate reporting requirements.
From a systemic perspective, linking the new technical requirements to data published by the EMA is consistent with the Agency’s role of ensuring the coherence and comparability of information on medicinal products at the Union level. Article 57 does not explicitly mandate the EMA to collect or share data on public financial support for research and development, but it does establish an institutional framework enabling the integration of various categories of information relating to the same medicinal product. The scope of this framework’s potential use for financial transparency purposes depends on detailed substantive provisions introduced in other legal acts and does not result directly from the technical changes under consideration in Article 26.
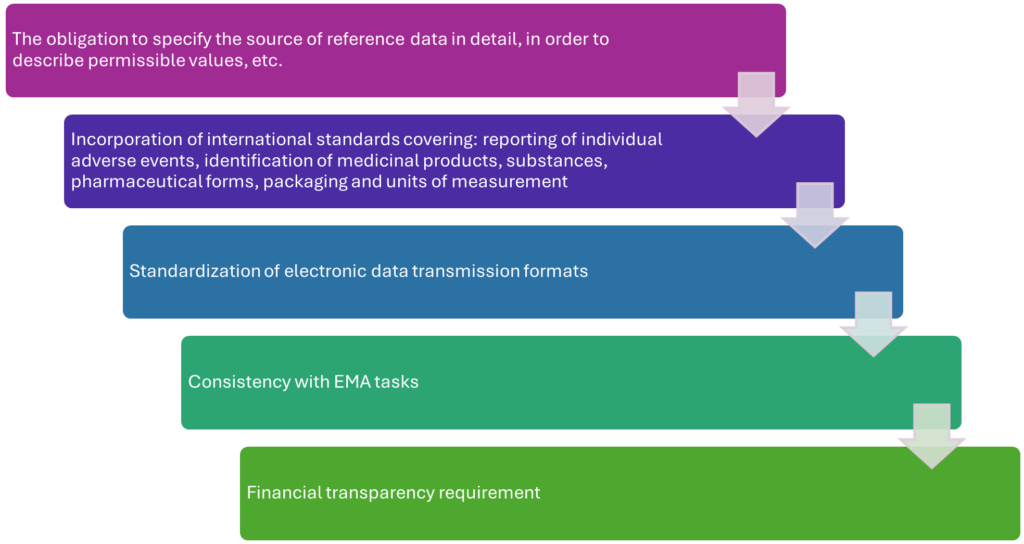
The changes introduced by Article 26 of the Implementing Regulation of 22 July 2025 amending Implementing Regulation (EU) No 520/2012 on pharmacovigilance activities provided for in Regulation (EC) No 726/2004 of the European Parliament and of the Council and in Directive 2001/83/EC of the European Parliament and of the Council generally include:
After a theoretical analysis, mainly devoted to the legal aspects of the changes and the consequences of the introduced regulation, it is worth paying attention to what practical consequences this may have, among others, within the scope of conducted activities.
One of the most obvious areas of impact of the new regulations is the activities of marketing authorization holders (MAHs). These are entities that apply for or obtain marketing authorization for a given medicinal product and are then responsible for monitoring its use after it is launched on the market.
Once the new regulations are implemented, MAHs will be required to develop and maintain dedicated websites for each product marketed in the EU. This requires implementing appropriate operational procedures and submitting reports to an external audit. Each website should include, among other things, a summary of the financial support granted for product-related research and development activities, even if these activities were conducted by external entities prior to the MAH’s involvement.
From a practical perspective, introducing such transparency could significantly impact the operations of pharmaceutical companies. Member States strive to increase the availability of medicinal products, and transparency in research funding and drug launches could strengthen the position of regulators and payers. Consequently, pharmaceutical companies may feel greater pressure during pricing and reimbursement negotiations, as information about financial support provided will be publicly available and could influence the assessment of drug launch costs.
In practice, this means not only additional administrative and auditing obligations for MAH, but also careful planning of communication and negotiation strategies to ensure compliance with regulations while maintaining competitiveness on the market.
As Member States strive to maintain and improve access to medicinal products and introduce transparency in the scope of support covering the use of financial resources for introducing medicinal products to the market, this regulation may increase the negotiating pressure on pharmaceutical companies through awareness and transparency of subsidies during price and reimbursement negotiations.
Full transparency may raise debates on issues related to the protection of trade secrets, know-how, and the protection of business secrets regulated, among others, by Article 39 of the TRIPS Agreement. Significant doubts may arise regarding the risk of replicating competitors’ R&D strategies, which may raise doubts as to whether the proposed regulations contain sufficient clauses ensuring the protection of business secrets.
The proposed solution appears to be significant given the systemic approach to the application of EU law in relation to Article 107 TFEU. It seems that such disclosure of financing could facilitate the identification of unlawful state aid, which could, among other things, facilitate proceedings before the European Commission.
The introduction of the obligation to disclose information is part of strengthening social responsibility and consumer awareness regarding the implementation of ESG (Environmental, Social, Governance) standards.
Funding transparency can be classified as a governance element that reveals the relationship between the public sector and commercial entities. From a societal perspective, this transparency demonstrates that innovation is not solely related to private pharmaceutical risk but also the result of the collaboration of public funds, academic infrastructure, and private capital.
In the social context, transparency of financing can influence the perception of the pharmaceutical industry by the public, patients, and public payers. Disclosure of subsidies can reinforce public expectations for appropriate pricing policies, while companies themselves may perceive such transparency as an element of their repudiation strategy and non-financial reporting.
At the international level, the issue of transparency in the financing of pharmaceutical innovation also appears in documents from the World Health Organization (WHO) and the OECD, which emphasize the importance of transparency in ensuring fair access to medicines and rationalizing pricing policies. Against this backdrop, the European Union’s approach can be viewed as more systemic and integrated with the regulatory lifecycle of medicinal products. At the same time, the question arises whether expanded disclosure obligations will lead to a situation of so-called “over-transparency,” where companies operating in the EU market will be subject to more stringent disclosure obligations than their competitors in other regions of the world, which may be significant for the global competitiveness of the European pharmaceutical sector.
A particularly concerning issue related to the proposed regulatory changes is the lack of clarified enforcement strategies and legal consequences related to the new requirements. In practice, it may be necessary to determine which body – the EMA, the European Commission, or the relevant national authorities – will be responsible for verifying the compliance of disclosed data with the actual state of R&D funding. The lack of clear regulations or sanctions regarding aspects of non-compliance with the obligation may weaken its practical significance. Regardless of the basis of formal legal sanctions, enforcement of the disclosure obligation may rely not only on legal instruments but also on market instruments or broadly understood social pressure.
The current legal regulations (de lege lata) provide the institutional and technical framework for collecting, standardizing, and publicly disclosing data on medicinal products, with the scope of this information remaining essentially limited to regulatory aspects related to the quality, safety, and efficacy of medicines. Article 57 of Regulation (EC) No 726/2004 provides a key legal basis for the EMA’s operation as a central information authority, but does not explicitly cover transparency obligations regarding research and development funding. From a de lege ferenda perspective, it can be argued that further development of transparency requirements, particularly regarding public financial support for R&D, requires explicit support in substantive EU law, while leveraging the EMA’s existing data infrastructure and experience gained from implementing the tasks specified in Article 57. Therefore, the direction of potential legal changes may lie not in redefining the Agency’s role, but rather in gradually expanding the catalog of information related to medicinal products, while maintaining systemic coherence and a clear division of responsibilities between the EMA, the Commission, and the Member States.
Sources:
1. Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the marketing authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency, OJ L 136, 30.04.2004, pp. 1–33.
2. Commission Implementing Regulation (EU) 2025/1466 of 22 July 2025 amending Implementing Regulation (EU) No 520/2012 on the performance of pharmacovigilance activities provided for in Regulation (EC) No 726/2004 and Directive 2001/83/EC, OJ EU L …, 23/07/2025.
3. Commission Implementing Regulation (EU) No 520/2012 of 19 June 2012 on the performance of pharmacovigilance activities provided for in Regulation (EC) No 726/2004 and Directive 2001/83/EC, OJ EU L 159, 20.06.2012, pp. 5–25.
4. European Commission, Proposal for a Regulation of the European Parliament and of the Council laying down Union marketing authorisation procedures for medicinal products for human use and the functioning of the European Medicines Agency, COM(2023) 193 final, EUR-Lex: Biznes.gov.pl, information on support for research and development activities and instruments for financing innovation, https://www.biznes.gov.pl/pl/portal/004268
5. European Medicines Agency, European Public Assessment Reports (EPAR), https://www.ema.europa.eu/en/medicines
6. Public contributions to R&D of medical innovations: A framework for analysis Claudia Wild * , Ozren Sehic , Louise Schmidt, Daniel Fabian, Austrian Institute for Health Technology Assessment (AIHTA), Vienna, Austria
7. Öffentliche Beiträge zur Arzneimittelentwicklung – Claudia Wild und Daniel Fabian